
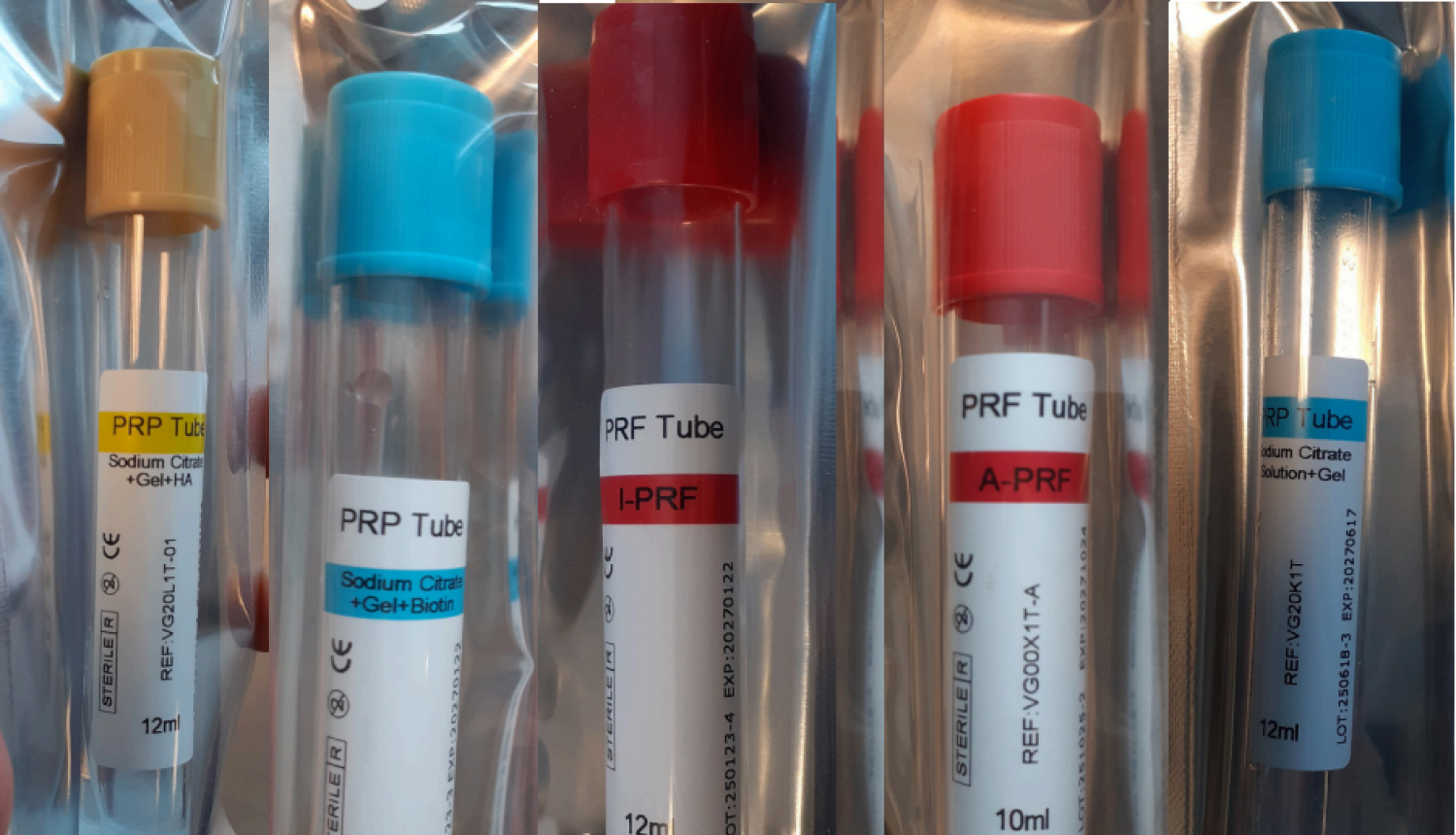
Veselības inspekcija, veicot medicīnisko ierīču tirgus uzraudzību, konstatēja, ka Latvijas komersants izplata medicīnisko ierīču prasībām[1] neatbilstošus asins savākšanas stobriņus, kurus piedāvā sekojošo procedūru veikšanai: PRP (Platelet-Rich Plasma) jeb trombocītiem bagātināta plazma un PRF (Platelet-Rich Fibrin) jeb trombocītiem bagātināts fibrīns.
PRP un PRF procedūru pamatā ir pacienta paša bioloģiskā materiāla izmantošana, lai stimulētu audu reģenerāciju, dzīšanu un atjaunošanos. Šīs ierīces plaši tiek izmantotas arī estētiskajā medicīnā.
PRP un PRF procedūrai paredzētie stobriņi būtiski atšķiras no stobriņiem, kas paredzēti laboratorijām, lai savāktu un uzglabātu asinis. Līdz ar to izmantojot prasībām neatbilstošus stobriņus PRP un PRF, ārstniecības personas riskē ne tikai ar neveiksmīgu procedūru, bet arī ar pacienta veselību.
Informācija pacientiem
Inspekcija vērš uzmanību, ka Pacientu tiesību likums nosaka pacienta tiesības saņemt saprotamu informāciju par savu ārstēšanu un veiktajām procedūrām, tostarp par ārstēšanas metodēm, kurās tiek izmantoti PRP vai PRF stobriņi. Ārstniecības personai ir pienākums pacientu informēt par iespējamām blakusparādībām, ko var izraisīt konkrētais materiāls vai tā apstrādes veids. Savukārt ārstniecības iestāžu vadītājiem un ārstniecības personām pirms PRP un PRF stobriņu iegādes ir pienākums pārliecināties, ka procedūrā izmantotie stobriņi ir prasībām atbilstošas medicīniskās ierīces un ka ražotājs tos ir paredzējis konkrētās procedūras veikšanai.
Prasības ārstniecības iestādēm un ārstniecības personām
Svarīgākās lietas, kam ārstniecības iestādēm un ārstniecības personām nepieciešams pievērst uzmanību pirms PRP un PRF iegādes:
-
Ja uz stobriņiem un iepakojuma norādīts, ka tā ir in vitro diagnostikas medicīniskā ierīce (IVD – In Vitro Diagnostic) vai “tikai laboratorijas lietošanai” (For laboratory use only), tas ir nopietns brīdinājums, ka šī ierīce ir paredzēta TIKAI asins paraugu savākšanai laboratorijā, nevis tam, lai pēc apstrādes asinis ievadītu atpakaļ cilvēka ķermenī, kā to paredz PRP un PRF procedūras. Šādos stobriņos var būt vielas, kuru drošība nav pārbaudīta situācijās, kad tās nonāk saskarē ar cilvēka audiem vai tiek ievadītas organismā. Ražotājam, kas piedāvā PRP un PRF procedūrai paredzētus stobriņus, ar klīniskiem pētījumiem ir jāpierāda, ka iegūtais koncentrāts tiešām veicina dzīšanu vai audu reģenerāciju, nevis ir tikai neaktīvs šķidrums, kā arī tas, ka tas ir drošs;
-
Ja stobriņš vai iepakojums ir marķēts ar CE zīmi, bet blakus nav norādīti četri cipari (piemēram, CE 2797), tad PRP un PRF stobriņu ražotājs nav veicis atbilstības novērtēšanas procedūru atbilstoši medicīnisko ierīču prasībām. Ražotājam, lai laistu tirgū PRP un PRF stobriņus, ir jāvienojas ar paziņoto struktūru par medicīnisko ierīču atbilstības novērtēšanas procedūru. Neatkarīga Eiropas Savienības paziņotā struktūra pēc novērtēšanas procedūras izsniedz EK sertifikātu, kas ļauj ražotājam ierīci marķēt ar CE atbilstības zīmi, kurai blakus būs norādīts paziņotās struktūras četru ciparu identifikācijas kods;
- Ja stobriņu etiķete ir pilnīgi neitrāla, bez ražotāja nosaukuma, partijas numura un derīguma termiņa, tad tā neatbilst medicīnisko ierīču prasībām. Katrai medicīniskajai ierīcei jābūt identificējamai pat tad, ja tā ir izņemta no tirdzniecības iepakojuma. Ārstniecības iestādēm ir jānodrošina medicīnisko ierīču izsekojamība (UDI, LOT) un tas ir svarīgi, ja rodas komplikācijas vai negadījumi un nepieciešams pārbaudīt konkrētās partijas kvalitāti. Latvijas normatīvie akti[2] paredz vigilances ziņojumu sistēmu un vigilances datubāzes uzturēšanu;
- Ja komersants PRP un PRF piedāvā par cenu, kas ir krietni zemāka nekā vidēji tirgū, tas rada aizdomas, ka ražotājs nav veicis atbilstības novērtēšanas procedūru, kas ir gan laikietilpīgs, gan samērā dārgs process;
- Ja PRP un PRF stobriņi, kā arī to izplatītājs nav reģistrēts medicīnisko ierīču datubāzē LATMED[3], tad šīs medicīnas ierīces statuss Latvijas tirgū nav leģitīms. Līdz ar to ārstniecības iestāde nedrīkst sniegt veselības aprūpes pakalpojumu ar šādu ierīci.
- Papildus ar informāciju par ražotāju un ierīci var iepazīties Eiropas Savienības medicīnisko ierīču datubāzē EUDAMED[4].
Ražotāju, importētāju un izplatītāju pienākumi
Savukārt importētāju un izplatītāju pienākumos ietilpst pārliecināties:
- vai ierīcei ir ražotāja izsniegta ES atbilstības deklarācija un paziņotās struktūras sertifikāts par atbilstību medicīnisko ierīču prasībām;
- vai uz ierīces etiķetes, tirdzniecības iepakojuma, lietošanas pamācības ir norādīts ierīces ražotājs, kas skaidri norāda kurš uzņemas atbildību par ierīci.
- vai uz ierīces etiķetes, tirdzniecības iepakojuma ir norādīta informācija: ka ierīce ir medicīniska ("Medical Device" vai simbols MD), sērijas numurs, UDI, datums līdz kuram ierīce ir droši lietojama un citas.
- vai ierīcei ir pievienota lietotājam paredzētā informācija uz etiķetes un lietošanas pamācībā (valsts valodā);
- vai uz ierīces etiķetes, tirdzniecības iepakojuma, lietošanas pamācībā ir norādīts ražotāja Eiropas Savienības pilnvarotais pārstāvis, ja ierīce ražota ārpus ES (piemēram, ASV, Ķīnā, Turcijā), kā arī vai ir pieejama informācija par importētāju.
Atbilstoši Ministru kabineta 2024. gada 15. augusta noteikumu Nr. 461 “Medicīnisko ierīču noteikumi” 9. un 29. punktam medicīnisko ierīču izplatītāji iesniedz Zāļu valsts aģentūrā iesniegumu, kurā norāda informāciju par sevi un medicīniskajām ierīcēm, kuras plānots izplatīt.
Veselības inspekcijas rīcība un turpmākie pasākumi
Veselības inspekcija, konstatējot prasībām neatbilstošus PRP un PRF stobriņus, rīkojās savas kompetences ietvaros un uzdeva komersantam:
- nekavējoties pārtraukt PRP un PRF izplatīšanu (tirdzniecību);
- veikt PRP un PRF stobriņu atsaukšanu no visiem klientiem.
Veselības inspekcija, kā medicīnisko ierīču tirgus uzraudzības iestāde savā tīmekļa vietnē publicē informāciju par neatbilstošām ierīcēm, lai novērstu sekas, kuras var radīt neatbilstošu preču piedāvāšana tirgū, un informētu sabiedrību, t.sk. ārstniecības iestādes un pacientus[5].
[1] Eiropas Parlamenta un Padomes 2017. gada 5. aprīļa Regula 2017/745, kas attiecas uz medicīniskām ierīcēm, ar ko groza Direktīvu 2001/83/EK, Regulu (EK) Nr. 178/2002 un Regulu (EK) Nr. 1223/2009 un atceļ Padomes Direktīvas 90/385/EK un 93/42/EEK
[5] Likums “Par atbilstības novērtēšanu” https://likumi.lv/ta/id/63836-par-atbilstibas-novertesanu