
Darbību joma: Veselības inspekcijas Medicīnisko ierīču uzraudzības nodaļa (turpmāk - Inspekcija) laika posmā no 22.12.2020. līdz 26.02.2021. veica 9 attālinātas kontroles uzņēmumos, kas Latvijas tirgū piedāvā in vitro diagnostikas (turpmāk - IVD) medicīniskās ierīces: SARS-CoV-2 antivielu un/vai antigēnu ātros testus (turpmāk - Testi).
Pārbaužu ietvaros Inspekcija izvērtēja Testus, kuri atbilstoši IVD medicīnisko ierīču prasībām[1] tiek klasificēti kā piederīgi pie kategorijas citi jeb other. Šīs kategorijas Testi ir paredzēti tikai profesionālai lietošanai veselības aprūpes speciālistam. SARS-CoV-2 ātrie testi, kas klasificēti kā pašpārbaudes ierīces, kuras ražotājs paredzējis lietošanai mājas apstākļos personai bez profesionālām zināšanām medicīnā pārbaudes laika posmā no 22.12.2020. līdz 26.02.2021. Latvijas tirgū netika piedāvāti.
Pārbaužu mērķis: izvērtēt Latvijas tirgū esošo Testu atbilstību IVD medicīnisko ierīču prasībām. Pārbaudes veiktas, jo
- pieaug iedzīvotāju un komersantu jautājumu un pieprasījumu skaits par SARS-CoV-2 testiem un to atbilstību;
- SARS-CoV-2 vīrusa izplatības ietekmē pieaug Testu pieprasījums un piedāvājums tirgū.
Kontroles objektu atlase: Informācija par Latvijas tirgū pieejamiem Testiem un uzņēmumiem, kas tos piedāvā, tika atlasīta veicot Latvijā reģistrēto uzņēmumu interneta vietņu satura analīzi.
Vērtēšanas kritēriji: Atlasītiem uzņēmumiem tika nosūtīts rakstisks Inspekcijas pieprasījums iesniegt informāciju par preces piegādātāju un gadījumā, ja uzņēmums preci Latvijā ieveda pats - Testa atbilstības un kvalitātes apliecinošo dokumentāciju:
- Ražotāja sastādītu EK atbilstības deklarāciju,
- lietošanas instrukcijas oriģinālu un tulkojumu valsts valodā,
- tirdzniecības iepakojuma un komponenšu fotoattēlus no visām iepakojuma un komponenšu pusēm tā, lai būtu attēlā redzams un identificējams ierīces ražotājs, modelis, sērijas numurs un CE marķējums.
Inspekcija uzņēmumus papildus informēja, ka prasības IVD medicīniskām ierīcēm ir noteiktas Noteikumos Nr.689, kur 9.punkts nosaka, ka medicīniskās ierīces var laist tirgū vai ieviest, ja:
9.1. medicīniskā ierīce, attiecīgi piegādāta un pienācīgi uzstādīta, uzturēta un izmantota atbilstoši paredzētajam mērķim, atbilst šajos noteikumos noteiktajām būtiskajām prasībām un tai ir veiktas šo noteikumu 9. nodaļā norādītās atbilstības novērtēšanas procedūras atbilstoši šo noteikumu 13. punktam;
9.2. ir sastādīta EK atbilstības deklarācija un ierīces ir marķētas ar CE atbilstības marķējumu, ar ko ražotājs apliecina, ka ierīce atbilst visām attiecināmajām normatīvo aktu prasībām.
Kontroles laikā Inspekcija vērtēja:
- lietošanas instrukcijas satura atbilstību Noteikumu Nr.689 107.punktam, kā arī valsts valodas lietošanas instrukcijas pieejamību kā to paredz Noteikumu Nr.689 25.punkts. Tika vērtēts vai tulkojums ir pilnībā atbilstošs instrukcijas oriģinālam;
- Testa tirdzniecības iepakojuma un tā komponenšu etiķešu satura atbilstību Noteikumu Nr.689 104.punkta prasībām;
- ražotāja sastādītās EK atbilstības deklarācijas pieejamību un CE marķējuma atbilstību Noteikumu Nr.689 9.2.punktam.
Rezultāti un to analīze: Kopumā Inspekcija apzināja 12 uzņēmumus. Izvērtējot uzņēmumu sniegtās atbildes, Inspekcija veica attālinātas kontroles 9 uzņēmumiem (izplatītāji un importētāji), kuri Latvijas tirgū piedāvā Testus.
Kontroles ietvaros vērtēti iepriekš minētie kritēriji un normatīvo aktu prasību izpilde.
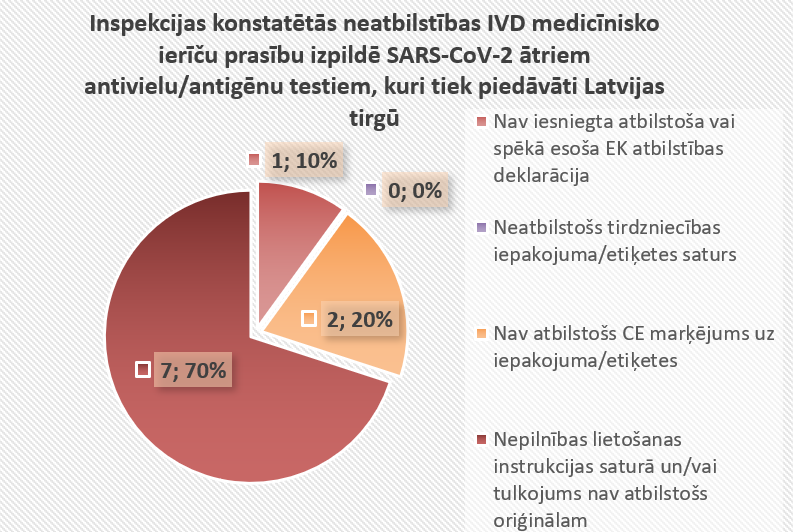
- Pārbaužu rezultāti liecina, ka no 9 kontrolētiem uzņēmumiem, pilnīgu atbilstību Noteikumu Nr.689 prasībām sākotnēji spēja nodrošināt 3 uzņēmumi jeb 33%.
- Konkrēto Testu tirdzniecības iepakojuma un komponenšu etiķešu satura analīzes rezultātā netika konstatēta Noteikumu Nr.689 prasību neizpilde.
- No kopējā neatbilstību skaita 70% gadījumos, Inspekcija konstatēja nepilnības ražotāja sastādītās lietošanas instrukcijas saturā vai lietošanas instrukcijas tulkojums valsts valodā nebija pilnvērtīgs oriģinālai versijai.
- 10% jeb 1 konstatētā neatbilstība ir pamatojama ar to, ka pēc pirmreizējā Inspekcijas pieprasījuma uzņēmums neiesniedza spēkā esošu un/vai ar Testu identificējamu EK atbilstības deklarāciju.
- 20% jeb 2 konstatētās neatbilstības ir saistītas ar prasībām neatbilstošu CE atbilstības marķējumu uz Testu komponentēm – paraugu savākšanas kociņiem. Šo kontroļu rezultātā konstatēts, ka Testos ievietotie paraugu savākšanas kociņi ir apzīmēti kā sterilas medicīniskas ierīces, toties blakus CE atbilstības marķējumam nav norādīts paziņotās iestādes, kas veikusi šo komponenšu atbilstībās novērtēšanas procedūru, četru ciparu identifikācijas kods. Līdz ar to Inspekcija secināja, ka šīs komponentes nav atbilstošas Noteikumu Nr.689 13.punktam un Testiem, kuru sastāvā ir iekļautas neatbilstošās komponentes, tika veikta preču turpmākas izplatīšanas apturēšana un uzņēmumiem uzdots informēt patērētājus un komersantus, kuriem Testi tika izplatīti, par konstatēto neatbilstību, kā arī uzdeva organizēt neatbilstošo Testu atsaukšanu no tirgus.
Inspekcijas turpmākās darbībās: Pārbaužu rezultātā Inspekcija sastādīja kontroles aktus un, attiecīgajos gadījumos – preču izplatības apturēšanas aktus, kuru ietvaros iepazīstināja uzņēmumus ar IVD medicīnisko ierīču normatīvo aktu prasībām un informēja par Inspekcijas konstatētām neatbilstībām.
Medicīnisko ierīču izplatītājiem vai importētājiem, kuru Testiem konstatēja neatbilstības, Inspekcija uzdeva veikt korektīvās darbības neatbilstību novēršanai. Inspekcija ir saņēmusi apliecinājumus no 9 uzņēmumiem jeb 100% par Inspekcijas uzdoto pasākumu izpildi.
Ar nolūku nodrošināt papildus informāciju par medicīnisko ierīču prasībām, kas ir attiecināmas uz Testiem un to komponentēm, ir sagatavots un Inspekcijas mājaslapā 20.01.2021. publicēts informatīvs raksts, pieejams: https://www.vi.gov.lv/lv/jaunums/informacija-par-covid-19-atro-antigenu-testiem.
Papildus Inspekcijas mājaslapas sadaļā: https://www.vi.gov.lv/lv/neatbilstosas-mediciniskas-ierices pastāvīgi tiek ievietota informācija par Latvijas tirgū konstatētiem, medicīnisko ierīču prasībām neatbilstošiem Testiem u.c. medicīniskām ierīcēm.
[1] Testu laišanu tirgū reglamentē Eiropas Parlamenta un Padomes Direktīva 98/79/EK (1998. gada 27. oktobris) par medicīnas ierīcēm, ko lieto in vitro diagnostikā (turpmāk – Direktīva 98/79). Direktīvas 98/79 prasības ir pārņemtas ar Ministru kabineta 2017. gada 28. novembra noteikumiem Nr. 689 „Medicīnisko ierīču reģistrācijas, atbilstības novērtēšanas, izplatīšanas, ekspluatācijas un tehniskās uzraudzības kārtība” (turpmāk – Noteikumi Nr.689).